🧬 Introduction
In humanitarian and forensic genetics, one of the most computationally tedious problems is also one of the most consequential: systematically comparing a set of reference family pedigrees against a set of candidate biological relatives to find which pairs are actually related to the same missing person. This is the core challenge of Missing Person Identification (MPI) and Disaster Victim Identification (DVI) workflows.
A defining constraint in all these scenarios is that the person of interest is not always genotyped. They may be deceased and unidentified, disappeared, suspected of theft and identity swap, or simply unwilling to provide a biological sample — or their remains may have been cremated or commingled in an ossuary, making individual sampling no longer possible. The analysis must therefore rely exclusively on the genotypes of their relatives — making indirect kinship inference the only available path to identification.
Most forensic geneticists working in this area are familiar with Familias3. It is an excellent, free, well-validated software platform for kinship evaluation — and it remains the standard for pedigree construction and allele frequency management in forensic genetics. What is not fully implemented in Familias3, however, is a mechanism for systematically running every MPI/DVI family against every POI Component (Person Of Interest Component) in a comparison workflow in an all-vs-all.
That gap is exactly what KinshipAssembly was designed to close.

⚖️ Statistical Hypothesis Testing between MPI/DVI families and POI Components
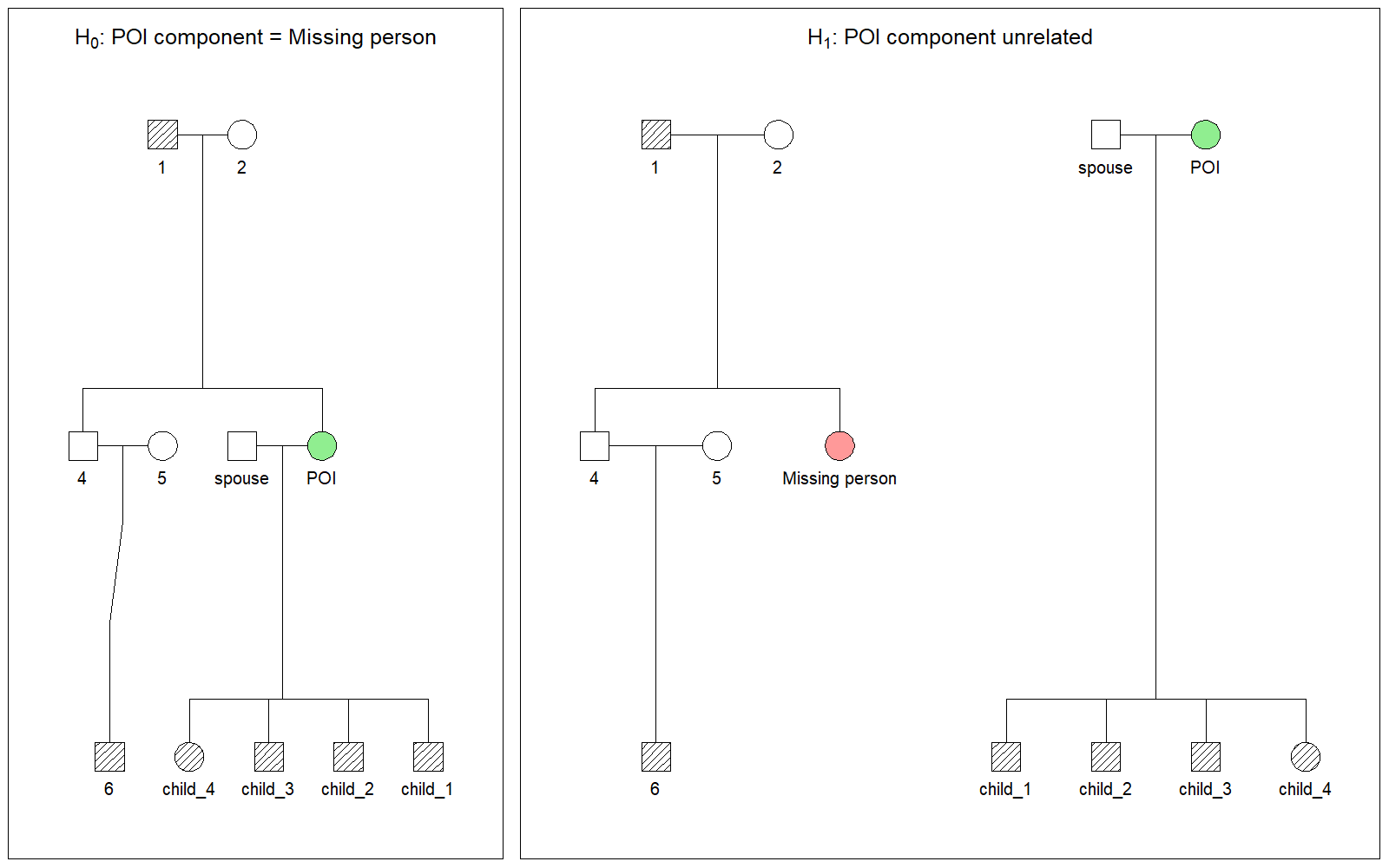
To understand why a dedicated application is necessary, it helps to consider the specific scenario KinshipAssembly is designed for — one where no direct reference profile is available on either side.
You work with two collections of pedigrees:
MPI/DVI families: families of missing or deceased individuals. Each pedigree contains typed reference relatives (parents, siblings, children) with an untyped node representing the missing person whose identity is being sought.
POI Components: family groups associated with a candidate. The POI themselves does not have a direct genotype profile available — what is available are the genotypes of their relatives. The “Component” reflects precisely this: kinship evidence assembled around a candidate who cannot or did not provide a sample directly.
The investigative question is: does this POI Component correspond to this missing person? Formally, this is evaluated via a Likelihood Ratio (LR):
$$ LR = \frac{P(\text{genetic data} \mid H_0)}{P(\text{genetic data} \mid H_1)} $$
where:
H₀ (relatedness hypothesis): the POI is the missing person. The two pedigrees are merged structurally — the untyped missing-person node and the untyped POI node are treated as the same individual. Neither has a direct genotype profile; the evidence comes entirely from the typed relatives on both sides, evaluated as a single connected pedigree.
H₁ (unrelatedness hypothesis): the POI is unrelated to the missing person. Both pedigrees remain independent, and the data are evaluated separately with no structural connection.
🔬 What KinshipAssembly Does
KinshipAssembly is a browser-based Shiny application (R) built around
forrel::kinshipLR() from the pedsuite ecosystem. Its core innovation is
workflow-level, not statistical.
The key design decisions for this application are:
📁 Dual-file architecture with Familias3 pedigrees
The app accepts two .fam files — one for the MPI/DVI collection and one for the
POI Components (both configured in the DVI/MPI module). This means you work entirely
within the Familias3 ecosystem for pedigree construction and allele frequency
management, then hand off to KinshipAssembly for the computation of the LR.
🔄 Mutation modelling built into the app
Three mutation models are available: None, Equal, and Extended stepwise (with configurable rate and range). This is particularly important when working with degraded samples or older pedigrees where mutation events are more likely.
⚙️ All-vs-all or targeted comparisons
You can choose between all-vs-all (every MPI/DVI family tested against every POI Component) or a targeted mode where you manually select specific pairs.
🎯 LR threshold filtering without recalculation
After running a comparison, you can adjust the LR threshold slider to hide pairs below any cutoff — without triggering a new computation.
🔎 Detailed Pairwise Analysis
Every row in the results table is clickable. Selecting a pair opens a detail panel showing:
- Side-by-side pedigree hypothesis plots (H₀ and H₁)
- A per-marker LR table with genotypes for all typed relatives
- Direct downloads for the pedigree image (PNG) and the per-marker data (CSV)
This is where the statistical work becomes interpretable.
🖥️ A Walkthrough with Real Output
A synthetic toy dataset with 15 MPI reference families and 20 POI Components with 24 STR markers was built to demonstrate the application’s functionality. The following screenshots show the application running on this dataset.
Loading data and configuring the run
Both .fam files are uploaded via the sidebar. The app confirms the pedigree count
for each file and activates the analysis controls. Here the mutation model is set to
None, the missing person label matches the untyped node in the pedigree, and the
comparison mode is All vs all. By default, the LR threshold is set to 1.
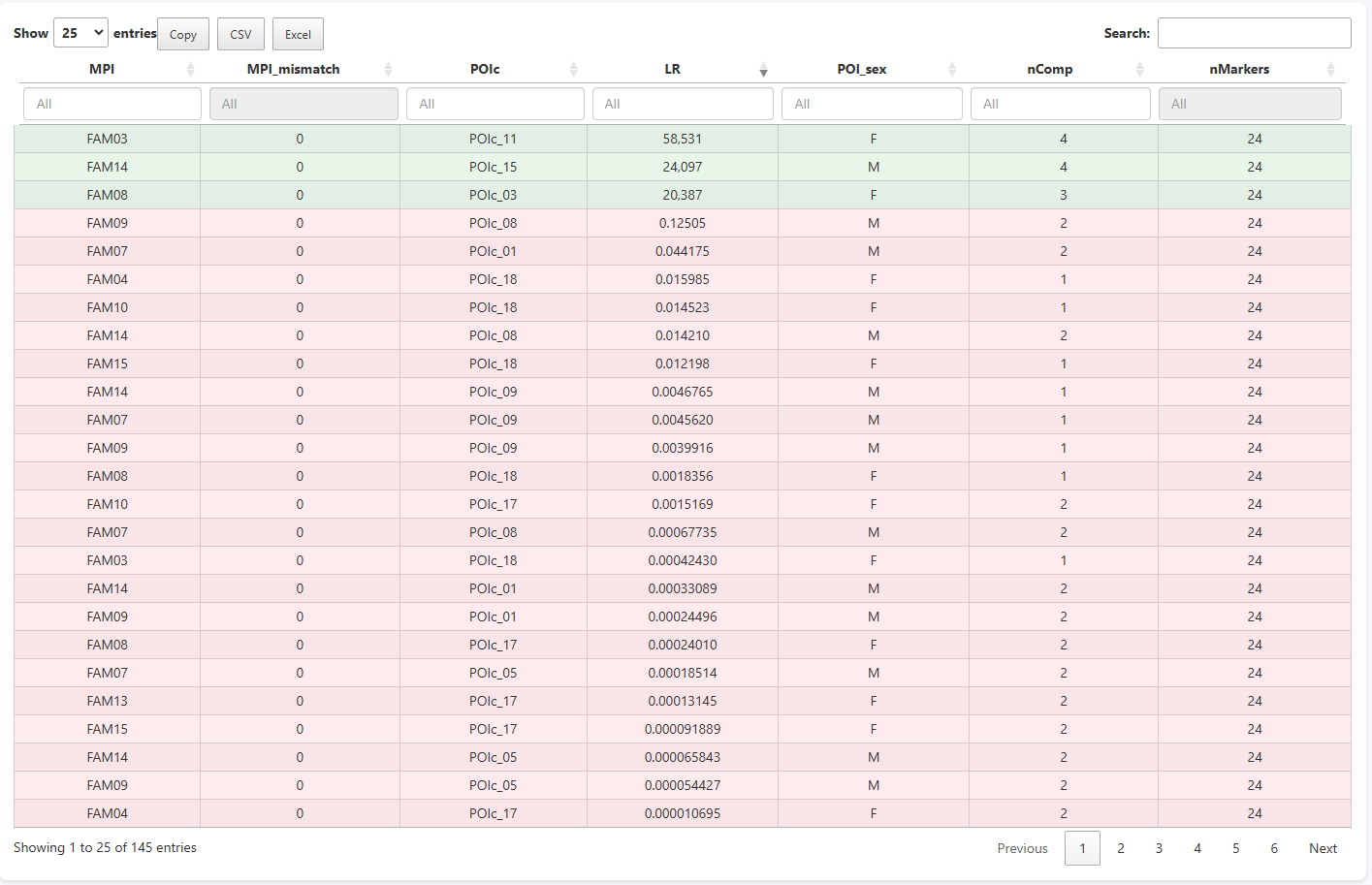
Results table after comparison
After clicking Compute LR, the Comparisons tab displays a table with one row per pair. Each row reports:
| Column | Meaning |
|---|---|
MPI | MPI/DVI family name |
MPI_mismatch | Mendelian inconsistencies within the MPI/DVI pedigree |
POIc | POI Component family name |
LR | Total likelihood ratio for this pair |
POI_sex | Recorded sex of the POI (M / F / UNK) |
nComp | Number of typed relatives in the POI Component |
nMarkers | Number of STR markers used after harmonisation |
Results are sorted by LR in descending order, immediately surfacing the most evidentially supported pairs. In this toy dataset, FAM03 vs POIc_11 tops the table with LR = 58.531 across 24 markers, with 4 typed relatives in the POI branch — a relatively strong result for a pedigree of this complexity.
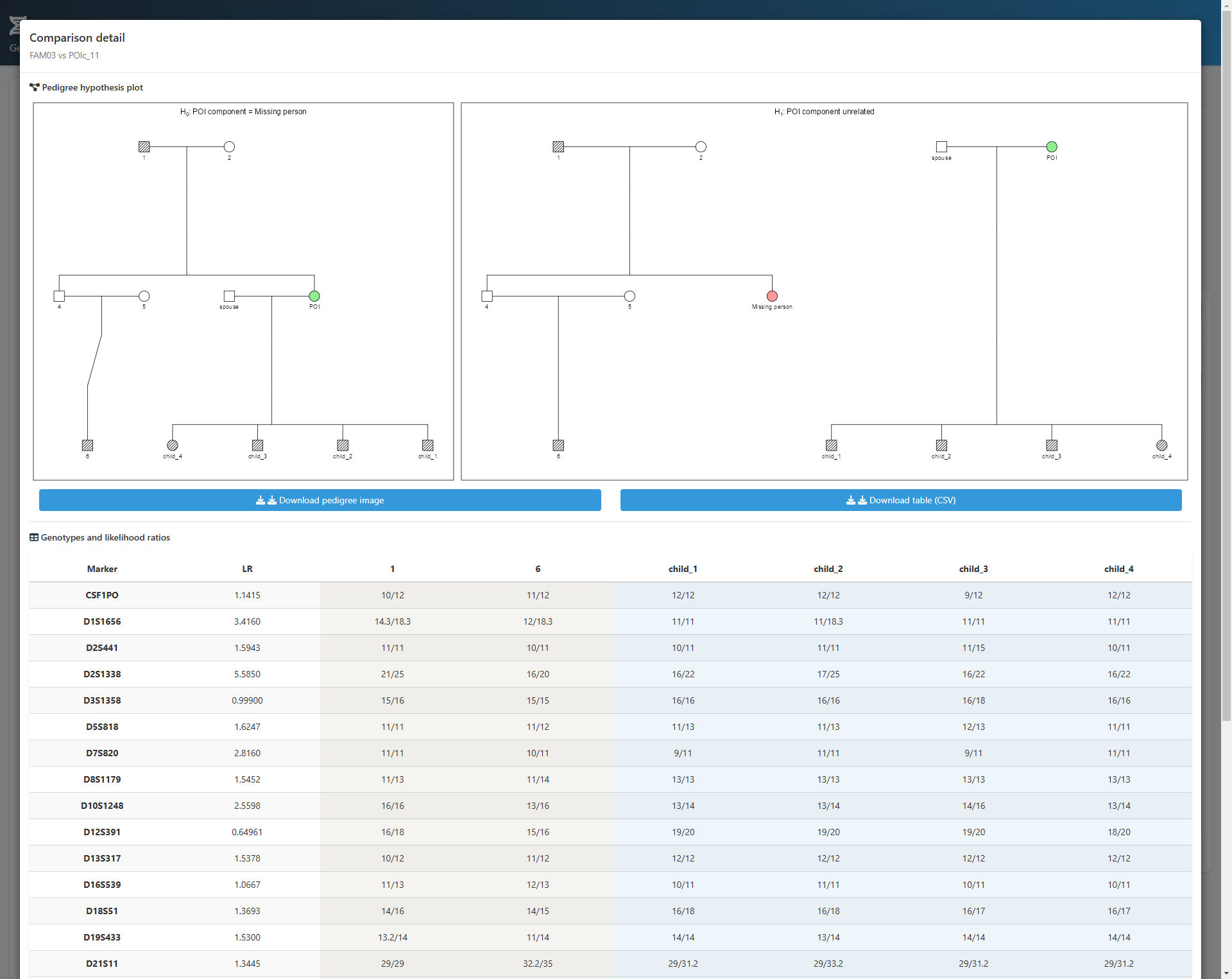
Comparison detail: pedigrees hypotheses and per-marker LRs
Clicking the FAM03 vs POIc_11 row opens the detail panel. On the left, the H₀ pedigree shows the merged structure — the POI is positioned as the missing person within FAM03. On the right, H₁ shows both families as completely independent units. Below the plots, a per-marker breakdown lists the LR contribution of each locus, alongside the genotypes of every typed individual.
This view is what makes the output verified: you can immediately see which markers are driving the LR upward (D2S1338 at 5.585, D7S820 at 2.816) and which are near-neutral (D3S1358 at 0.999), and verify genotype consistency.
🌍 When Is This Tool Relevant?
KinshipAssembly is designed for cases where families need to be reunified but no direct biological sample is available from the person being sought — and where the scale of the investigation makes manual one-by-one comparisons impractical. It is most applicable in:
Mass grave investigations: forensic teams working on mass grave cases typically have two growing collections — reference families reporting a missing person, and POI Components assembled from candidate individuals found at or near the site. Neither the missing person nor the candidate has a direct genotype available; identification must go through their relatives on both sides, and the number of combinations to evaluate quickly becomes unmanageable without automated processing.
Post-disaster investigations: aircraft accidents, natural disasters, or conflict events generate many unidentified victims and many reference families simultaneously. On one side, families reporting a missing person form the MPI collection; on the other, POI Components are assembled from candidates whose relatives have come forward. In both cases no direct profile is available — only the genotypes of relatives — and the volume of cross-comparisons quickly requires automated processing.
Identity substitution investigations: cases where a living individual is suspected of having assumed another person’s identity. The MPI family is the family of the person whose identity may have been taken; the POI Component is assembled from the relatives of the individual under suspicion — who may be alive but unwilling to provide a biological sample, or may be deceased. Neither side provides a direct profile from the person at the centre of the case, and a systematic all-vs-all comparison is needed to evaluate every possible pairing.
🛠️ Technical Stack
KinshipAssembly is built entirely in R using the following packages:
- Computation:
forrel::kinshipLR,pedtools,pedFamilias,pedmut - Interface:
shiny,shinyjs,shinyWidgets,DT - Data manipulation:
dplyr,purrr,tibble,stringr
The application requires R ≥ 4.5.0 and accepts .fam files generated by Familias3
(available at familias.no). The upload limit is set to
100 MB per request to accommodate large pedigree collections.
The full source code is available on GitHub:
git clone https://github.com/sbiagini0/KinshipAssembly.git
💡 Concluding Remarks
The statistical foundations of forensic kinship analysis — Bayesian likelihood ratios, mutation models, pedigree-aware genotype probability calculations — are well-established and implemented in pedigree-based packages. Building on these foundations, KinshipAssembly provides a workflow that systematically compares MPI/DVI families with POI Components in an all-vs-all comparison at large scale.
It is built around the fundamental reality of these cases: the person of interest cannot provide a sample, so identification must be achieved entirely through indirect kinship inference from relatives on both sides. It does not introduce new statistical methods; it makes existing, validated methods accessible at the scale that real-world humanitarian and forensic investigations require, through a browser-based interface that integrates directly with the Familias3 pedigree format that most forensic geneticists already use.
📚 References
- Vigeland, M. D. (2020). Pedigree Analysis in R. Academic Press. https://doi.org/10.1016/C2020-0-01956-0
- Familias3 software: https://familias.no/